
![]()
subsamplebam
Hopefully will allow you to randomly subsample a bam to get closer to even depth across the genome
Here are some quick comparisons:
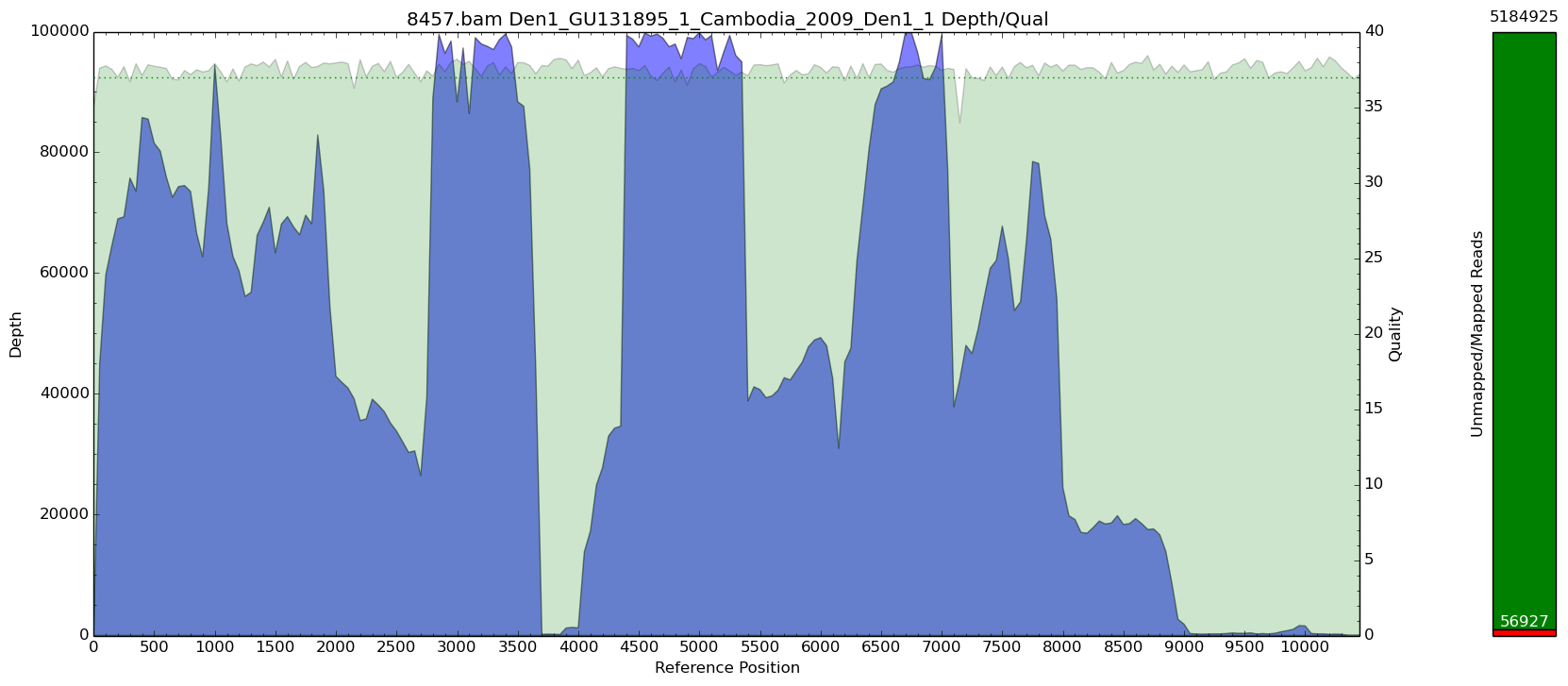
Original Sample
Here you can see that the read depth can vary drastically which may not be desired. There are some pretty massive peaks of coverage which drop off terribly.
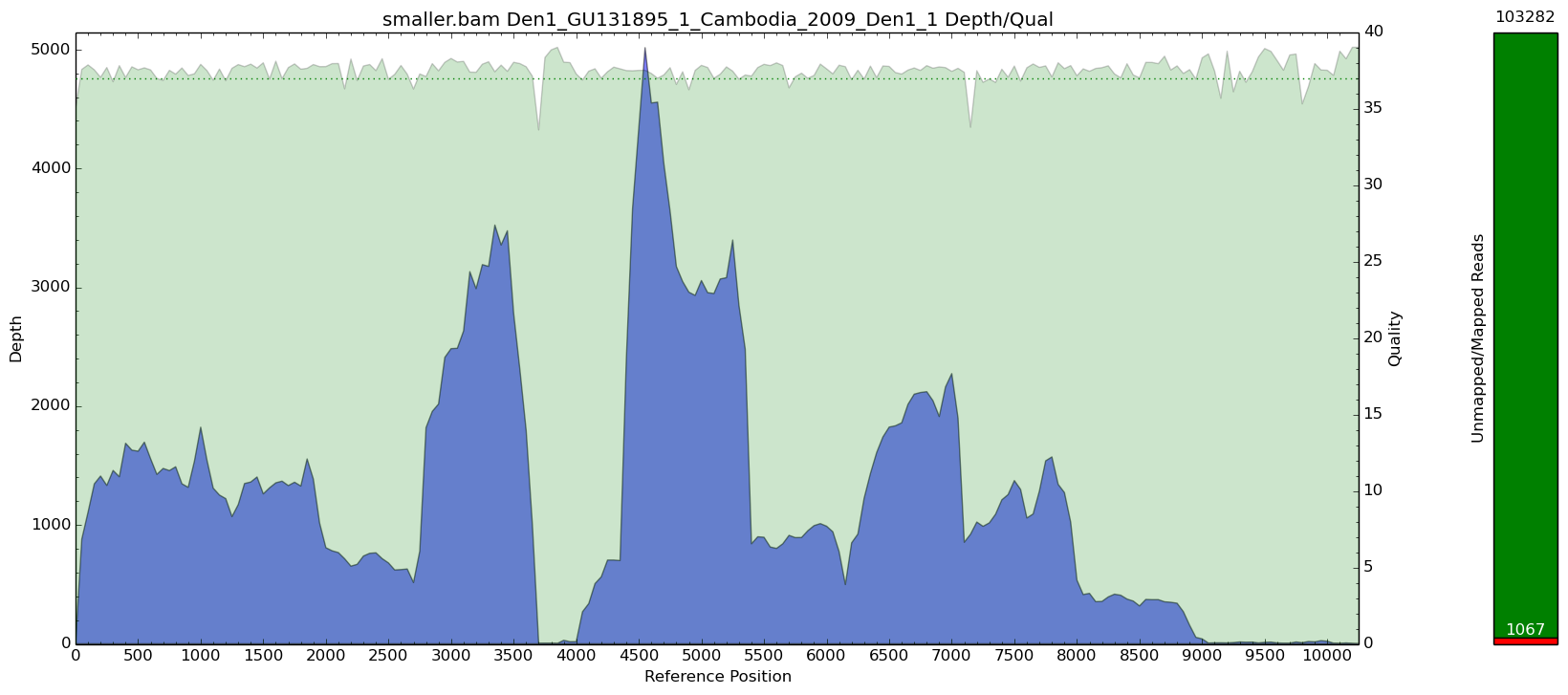
Using samtools to get a subsample
Here we have a bam file called 8457 which has over 5 million reads in it. I picked 1.02 to try and get about 100k of those reads and see what the coverage looks like after.
$> samtools view -hb -s 1.02 8457.bam > smaller.bam; samtools index smaller.bam
Here you can see that the peaks are not as drastic, but we also end up losing those low coverage regions as they get subsampled as well. Essentially, you just have the same issue on a smaller scale.
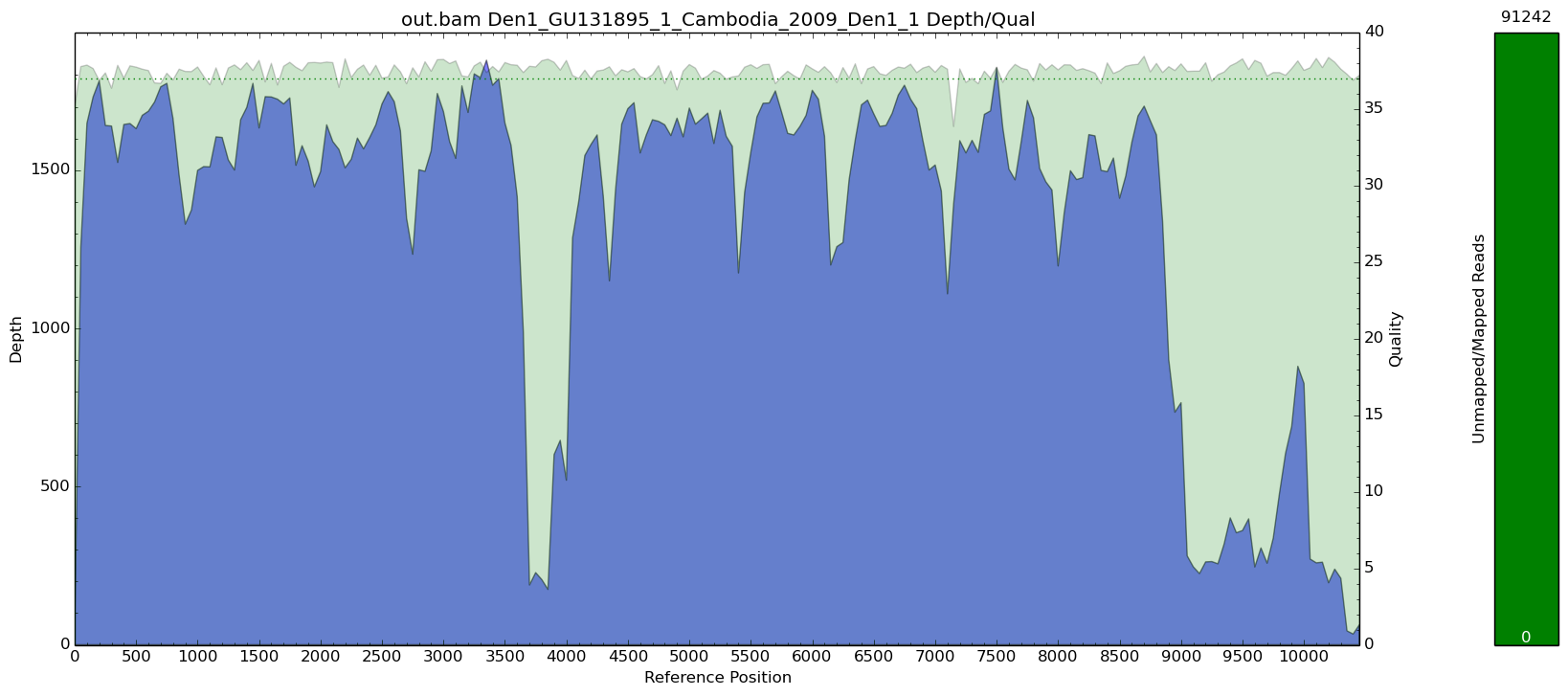
Using subsamplebam
With subsamplebam you can specify your --subsample wanted depth and it will grab that many random reads from each position on the genome which creates a more
uniform coverage.
$> samtools view -H 8457.bam
@HD VN:1.3 SO:coordinate
@SQ SN:Den1/GU131895_1/Cambodia/2009/Den1_1 LN:10474
@RG ID:Roche454 SM:8457 CN:None PL:L454
@RG ID:IonTorrent SM:8457 CN:None PL:IONTORRENT
@RG ID:MiSeq SM:8457 CN:None PL:ILLUMINA
@RG ID:Sanger SM:8457 CN:None PL:CAPILLARY
$> subsamplebam 8457.bam Den1/GU131895_1/Cambodia/2009/Den1_1:1-10747 --subsample 10 | samtools view -hSb - | samtools sort - subsampled; samtools index subsampled.bam;
You can see that the peaks still exist, but you end up with a much more uniform depth across than just doing random sampling with samtools.
Understanding the output
subsamplebam outputs to both stderr and stdout. The output to stderr will be the correct samtools region it is working on as well as sometimes you may see a line about Depth being only a certain amount.
Example output:
samtools view smaller.bam Den1/GU131895_1/Cambodia/2009/Den1_1:4600-4600
samtools view smaller.bam Den1/GU131895_1/Cambodia/2009/Den1_1:5744-5744
Depth for Den1/GU131895_1/Cambodia/2009/Den1_1:3693-3693 is only 4
samtools view smaller.bam Den1/GU131895_1/Cambodia/2009/Den1_1:3694-3694
samtools view smaller.bam Den1/GU131895_1/Cambodia/2009/Den1_1:4335-4335
samtools view smaller.bam Den1/GU131895_1/Cambodia/2009/Den1_1:5467-5467
The lines that begin with samtools are just showing you what position is being evaluated
The lines that begin with Depth are telling you that at that location, the read depth was only X which was lower than what you specified with --subsample
Installing samtools
If you don't already have samtools installed and in your PATH you can easily install it into this project as follows
tests/install_samtools.sh
export PATH=$PATH:$PWD/bin
This will only work for your current terminal session unless you modify your PATH in your .bashrc
Notes and TODO
Currently the way reads are selected means that even though you only specify say --subsample 10, you might end up with a great many reads covering a given area.
You can see this in the examples above that even though --subsample 10 was used, the coverage was well over 1500 for most of the genome. This is because when random reads are selected,
they may cover many base positions. So if your average read depth is 150 and you are sub selecting at 10 depth, the first base will contain 10 depth, but position 2 will contain those 10 reads, plus potentially 10 more reads. Then the 3rd position will
contain 10+10+10, and so on.
TODO
- Fix the issue noted above